
Quantum machine learning in drug discovery: Bridging quantum computing and pharmaceutical innovation

The integration of quantum computing and artificial intelligence, known as Quantum Machine Learning (QML), is emerging as a transformative approach capable of addressing significant computational challenges in pharmaceutical research. By leveraging the principles of quantum mechanics, QML holds potential for advancing drug discovery processes, as detailed in a recent comprehensive review by Smaldone and coauthors (Chemical Reviews, 2025).
In this post, I will explore critical insights from the review, outlining the theoretical foundations, methodologies, potential applications, current limitations, and future directions of QML, particularly focusing on Quantum Neural Networks (QNNs) and their promising roles in drug discovery.
Theoretical foundations of Quantum Machine Learning
Quantum Machine Learning synergistically combines quantum computation, which exploits quantum bits (qubits) exhibiting superposition and entanglement, with machine learning algorithms. QML algorithms can theoretically analyze and interpret complex molecular and biological datasets more efficiently than classical approaches.
Quantum Neural Networks (QNNs) are particularly emphasized, as they leverage variational quantum circuits—quantum analogs of classical neural network layers—to learn complex patterns within quantum states. These quantum circuits can potentially capture correlations and patterns in molecular data beyond the reach of classical neural networks, due to quantum parallelism and entanglement.
Why quantum methods in drug discovery?
Classical drug discovery workflows encounter bottlenecks arising from immense chemical complexity, the exponential growth of molecular search spaces, and the inherent computational limitations of classical hardware. Quantum computing offers methods to alleviate these issues by enabling:
Quantum parallelism: Efficient exploration of vast search spaces simultaneously due to superposition.
Enhanced molecular property predictions: Utilizing quantum methods to precisely predict molecular energies, conformational stabilities, and interaction profiles.
Quantum-enhanced molecular generation: Creating novel molecular structures with desired therapeutic properties through quantum generative models.
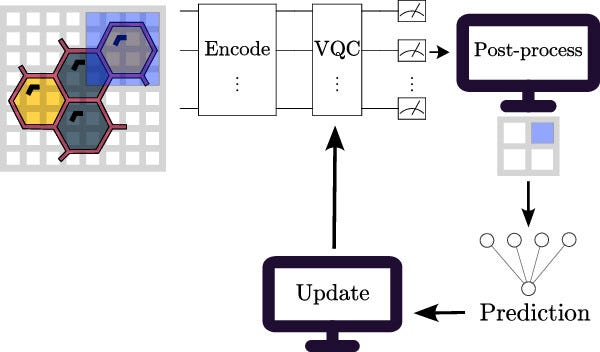
Technical implementation: How quantum neural networks work
QNN implementations typically include three fundamental steps:
1. Quantum data encoding
Classical molecular information (such as structural descriptors or chemical fingerprints) must first be encoded into quantum states. The review identifies three prominent encoding strategies:
Basis encoding: Encoding binary molecular data directly into quantum computational basis states.
Angle encoding: Real-valued molecular descriptors are encoded through quantum rotational gates (e.g., Rx, Ry, Rz rotations on Bloch spheres).
Amplitude encoding: High-dimensional data encoded into amplitudes of quantum states, offering exponential data compression, significantly enhancing computational efficiency and reducing required quantum resources.
2. Variational quantum circuits
Variational quantum circuits (VQCs) serve as quantum analogs of classical neural layers. These parametrized circuits utilize quantum gates to model complex, high-dimensional data correlations. Parameters of these circuits are optimized iteratively through hybrid quantum-classical algorithms, which employ classical optimization techniques (e.g., gradient descent, parameter-shift rules) to refine quantum state approximations.
3. Quantum measurement and classical optimization
The output from QNNs, obtained through quantum measurements, is classically post-processed and iteratively optimized. Variational quantum circuits rely on expectation-value measurements, computed via observables, which quantify molecular properties, binding affinities, or drug toxicities relevant to pharmaceutical tasks.
Cutting-edge Quantum Machine Learning applications in drug discovery
The review by Smaldone and coauthors provides detailed insights into three specific QNN-based methods showing particular promise in pharmaceutical research:
Quantum Graph Neural Networks (QGNNs)
QGNNs model molecular structures as graphs where nodes represent atoms and edges represent bonds. Quantum implementations have demonstrated enhanced predictive capabilities for molecular stability, chemical reactivity, and drug-target interactions compared to classical graph neural networks. Additionally, hybrid quantum-classical graph convolutional networks have already shown competitive performance in predicting protein-ligand binding affinities, indicating potential scalability and generalization improvements.
Quantum Convolutional Neural Networks (QCNNs)
QCNNs offer advantages by quantum processing small regions of molecular data—analogous to classical convolutional kernels—but with fewer parameters and reduced computational complexity. Such architectures have shown improvements in predicting molecular properties such as protein-ligand affinities and drug toxicities. Hybrid Quantum Convolutional Neural Networks (HQCNNs), which integrate classical convolutional layers with quantum circuits, have further demonstrated practical efficiencies in training time and resource usage.
Quantum Generative Adversarial Networks (QGANs)
Quantum GANs use quantum generative circuits to produce novel molecular structures with tailored properties. Despite facing challenges in generating fully chemically valid structures initially, recent advancements have highlighted that hybrid QGANs may improve drug-likeness and pharmacokinetic properties compared to purely classical generative approaches.
Challenges in Quantum Machine Learning implementation
Despite promising theoretical capabilities, practical implementation of quantum machine learning faces several significant challenges:
Quantum hardware limitations: Current quantum devices are limited by coherence time, qubit quality, and error rates (quantum noise), restricting the complexity and depth of quantum circuits that can be executed reliably.
Quantum algorithmic complexity: The complexity of quantum state preparation (particularly amplitude encoding) and optimization landscapes with barren plateaus (vanishing gradients in quantum circuits) present barriers to efficient training and convergence.
Optimization algorithms: Quantum weight updates remain computationally costly, as standard parameter-shift gradient methods have quadratic complexity concerning the number of circuit parameters. New, efficient optimization algorithms remain an active area of research.
Advances in quantum simulation tools
Addressing these challenges, advanced quantum simulation platforms such as NVIDIA’s CUDA-Q enable extensive simulation and validation of complex quantum circuits and hybrid algorithms, even in the absence of mature quantum hardware. GPU-accelerated quantum simulations facilitate rapid prototyping, scalability assessments, and development of robust quantum machine learning models suitable for pharmaceutical applications.
Future directions: Quantum Transformers and Bosonic Quantum Computing
Emerging quantum architectures such as Quantum Transformers could redefine generative models in chemistry. By leveraging quantum implementations of the self-attention mechanism—central to transformer architectures—researchers may uncover novel molecular structures inaccessible to classical computational methods.
Additionally, hybrid quantum processors integrating qubits with continuous-variable bosonic quantum modes (qumodes) represent an innovative computational paradigm that could significantly enhance data encoding efficiency, quantum circuit expressivity, and computational scalability.
Conclusion and outlook
Quantum machine learning represents a significant evolution in computational approaches to pharmaceutical sciences. While substantial hurdles remain, ongoing research and technological advancements continue to accelerate progress towards practical implementations. The promise of quantum computing, particularly in combination with machine learning methods, extends well beyond incremental improvements. It potentially signifies a foundational shift in drug discovery methodologies, enabling new avenues for addressing complex pharmaceutical challenges that classical computation alone may never fully resolve.