
Inverse protein folding with MapDiff: Combining diffusion models and deep learning for protein design

Protein design—the inverse problem of predicting protein structure—poses a deceptively challenging question: Given a desired three-dimensional backbone, can we reliably engineer an amino-acid sequence that adopts precisely that shape? This capability profoundly impacts fields such as drug discovery, enzyme engineering, and synthetic biology. Yet the complexity of protein interactions—especially in flexible loops, intrinsically disordered segments, and long-range contact networks—makes the inverse problem significantly more elusive than forward structure prediction.
In recent years, remarkable progress was achieved in forward prediction, culminating in DeepMind’s AlphaFold, which revolutionized our ability to infer structure directly from sequence. However, the reverse—sequence design given a structure—has proven more resistant. Classical physics-based optimization methods face computational bottlenecks and inaccuracies, while deep-learning approaches often struggle with propagating long-range context and reliably estimating uncertainty. Current state-of-the-art methods, though sophisticated, frequently falter in predicting flexible, disordered, or short-chain regions that defy straightforward structural inference.
Novelty of MapDiff
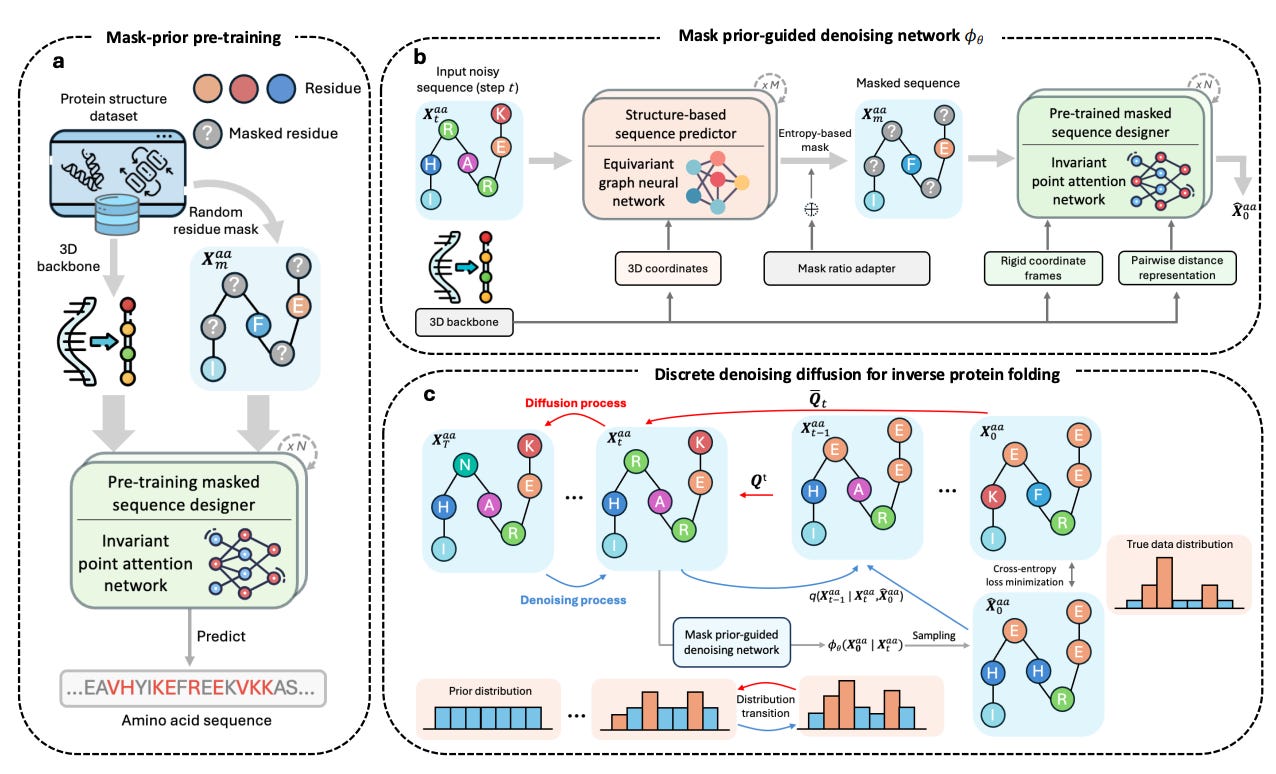
In their recent article published in Nature Machine Intelligence (May 2025), Bai and colleagues propose MapDiff, a mask-prior-guided denoising diffusion framework explicitly tailored to address inverse protein folding. This model recasts the sequence-design challenge as an iterative stochastic refinement process, starting from random “noise” sequences and progressively transforming them into biologically plausible ones. MapDiff introduces three core methodological innovations:
Global-aware equivariant graph denoiser:
An SE(3)-equivariant graph neural network (EGNN) preserves geometric invariances. Crucially, the network integrates global context, allowing each residue to incorporate both local geometry and long-range interactions throughout the protein.Mask-prior refinement:
Residues with high predictive entropy (uncertain predictions) are masked and re-evaluated by a pretrained Invariant Point Attention (IPA) module, significantly enhancing accuracy in structurally ambiguous regions.Efficient, uncertainty-aware sampling:
MapDiff employs discrete DDIM sampling, accelerating diffusion (from ~500 down to roughly 100 steps), while Monte-Carlo dropout ensembles mitigate overconfident predictions, enhancing robustness and practical utility.
Key insights and experimental validation
The authors rigorously evaluated MapDiff on several challenging benchmarks—CATH 4.2, CATH 4.3, TS50, and the time-split PDB2022 dataset. MapDiff achieves a median exact-residue recovery rate of approximately 61% on CATH datasets, surpassing prior baselines by 7–8 percentage points. Notably, improvements are most pronounced in traditionally challenging scenarios—short proteins (<100 residues), intrinsically disordered loops, and flexible secondary structures.
Critically, sequences generated by MapDiff, when refolded using AlphaFold2, yield median RMSD values as low as 2.9 Å and average pLDDT scores above 90. This demonstrates convincingly that better sequence accuracy directly translates into near-native 3-D structural fidelity in silico.
Real-world implications
From a practical standpoint, MapDiff provides tangible benefits beyond academic interest. In drug discovery, the method’s robustness in handling flexible regions allows researchers to confidently redesign catalytic pockets and binding interfaces, significantly increasing the likelihood of experimental success. Similarly, in synthetic biology and biocatalysis, MapDiff’s proficiency in predicting disordered segments substantially reduces the experimental risks associated with creating novel scaffolds for harsh industrial conditions.
Methodologically, MapDiff represents a significant leap by introducing diffusion modeling—previously transformative in image generation and natural-language processing—into protein design, without reliance on external evolutionary data or language-model pretraining. This approach potentially democratizes protein design, lowering entry barriers for research groups with less computational or experimental infrastructure.
Remaining challenges and future opportunities
Despite promising results, several open challenges remain. Rigorous wet-lab validation is essential: systematic expression, purification, and biophysical characterization of designed proteins will clarify how diffusion-model uncertainties map onto real-world experimental outcomes.
Another exciting direction is integrating evolutionary couplings captured by large protein language models, such as ESM-2. Leveraging these latent representations could enhance mutational robustness and evolutionary plausibility of designs. Furthermore, incorporating explicit physics-based constraints into the denoising process—penalizing steric clashes and unrealistic torsion angles early—could produce sequences more reliably suited for experimental validation.
Introducing an adaptive timestep controller that dynamically adjusts diffusion schedules based on structural complexity or ambiguity could improve computational efficiency and accuracy. Finally, extending the model beyond single-chain proteins to multimeric complexes, employing inter-chain attention, would unlock designs for sophisticated assemblies like therapeutic antibodies, enzyme complexes, and nanostructured biomaterials.
Summing up
MapDiff demonstrates that discrete denoising diffusion, combined with geometric equivariance and uncertainty-driven refinement, transforms inverse protein folding from a niche computational challenge into a practical generative task. By marrying rigorous structural reasoning with flexible probabilistic inference, MapDiff offers researchers a powerful tool for rational protein design—moving us significantly closer to routine creation of functional proteins that reliably translate from computational models into real-world laboratories.