
Accelerating chemical reaction optimization with high-throughput automation and machine intelligence

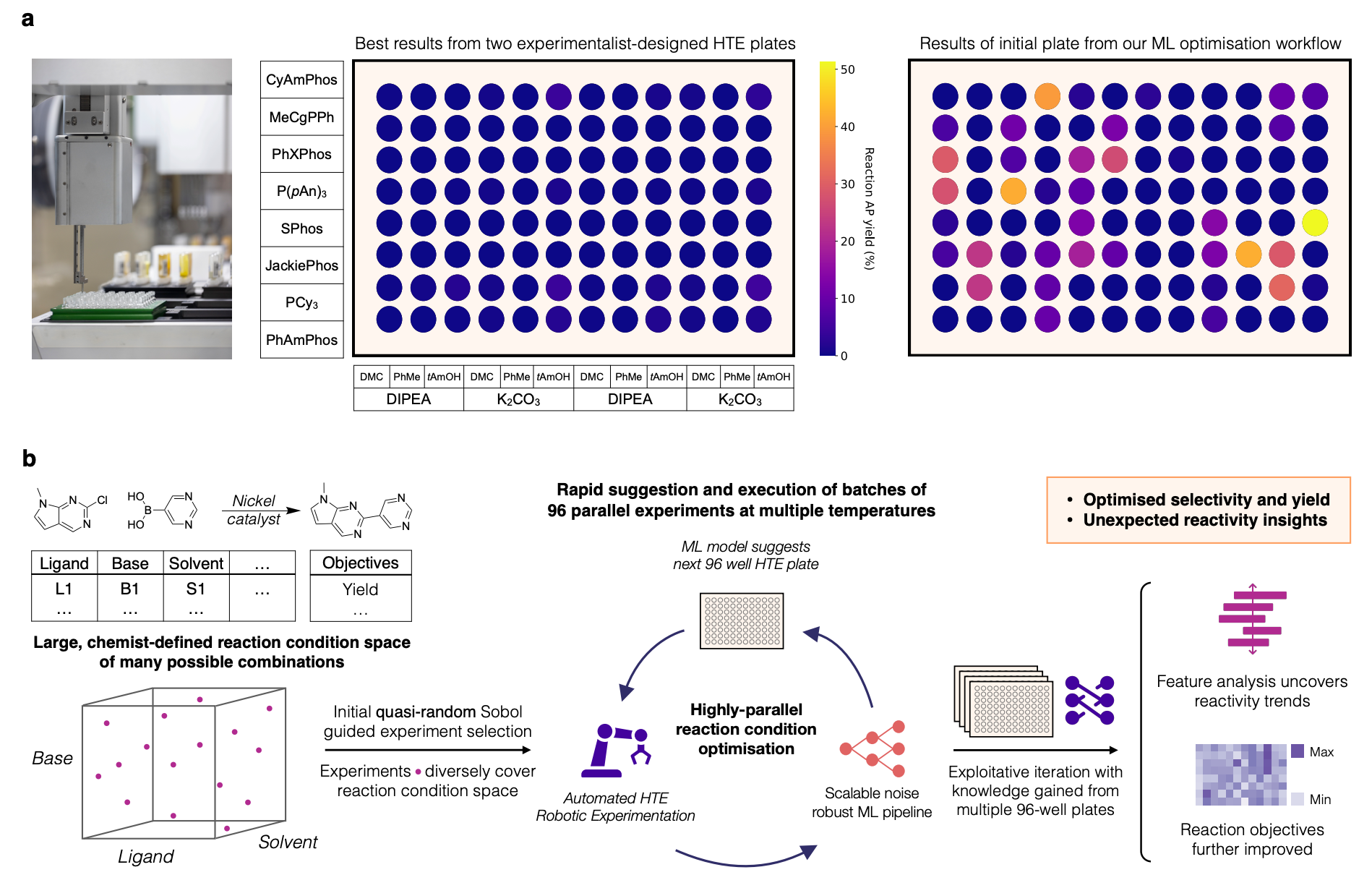
Chemical reaction optimisation is central to synthetic chemistry, underpinning a wide range of critical activities, from pharmaceutical drug development to the large-scale production of complex molecules. Typically, optimising chemical reactions involves systematically exploring different reaction parameters, including catalysts, solvents, reagents, and reaction conditions such as temperature and pressure. Traditional optimisation strategies, however, rely heavily on chemist intuition, manual experimentation, and one-factor-at-a-time (OFAT) approaches. These methods can be time-consuming, labor-intensive, and often fail to comprehensively explore the large combinatorial space of possible reaction conditions, especially as the complexity and dimensionality of the reaction space increases.
Recent advancements in automation and high-throughput experimentation (HTE) have significantly accelerated experimental workflows, enabling researchers to conduct many parallel reactions simultaneously. HTE approaches utilise robotic platforms to rapidly screen numerous reaction conditions, greatly expanding the range of feasible experiments. Nonetheless, even with automation, exhaustive experimentation is impractical for large reaction spaces. Therefore, effective methods are needed to guide experimental design, ensuring efficient exploration and optimisation of reactions without incurring unnecessary experimental overhead.
To address these challenges, data-driven approaches leveraging machine learning (ML) have emerged as promising alternatives. Among these, Bayesian optimisation—a technique that iteratively selects experiments based on predictive uncertainty and expected improvement—has shown considerable promise in chemistry. Despite their potential, existing ML-driven methods for chemical optimisation typically focus on small-scale batch experiments and face limitations when scaling up to the highly parallel batches typical in modern automated HTE setups. There is thus a critical need for scalable machine learning frameworks capable of efficiently handling high-dimensional reaction spaces and large parallel experimental batches, while maintaining robust performance and interpretability.
New research: Minerva, a scalable ML-driven framework for reaction optimization
In their recent publication, Joshua W. Sin and coauthors introduce Minerva, a highly scalable machine learning-driven framework designed specifically for automated multi-objective reaction optimization. Minerva integrates Bayesian optimisation, Gaussian process regression, and deep learning methodologies, and systematically leverages robotic high-throughput experimentation (HTE) to accelerate chemical reaction optimisation.
Minerva's optimization workflow begins with defining a comprehensive reaction search space. This involves selecting a broad set of plausible reaction conditions (such as catalysts, ligands, solvents, additives, and reaction temperatures) guided by expert chemical intuition and practical considerations. The initial experiments are then selected algorithmically using Sobol quasi-random sampling, ensuring diverse coverage of the defined reaction space.
Following these initial reactions, a Gaussian Process (GP) surrogate model is trained to predict reaction outcomes (such as yield and selectivity) across the entire search space. Subsequent experiments are then selected through an iterative Bayesian optimization procedure, specifically using scalable multi-objective acquisition functions such as q-NEHVI (q-Noisy Expected Hypervolume Improvement), which efficiently balances exploration and exploitation of the reaction space.
To accommodate practical constraints often encountered in experimental workflows, such as restrictions on the number of unique reaction temperatures per batch, Minerva incorporates constraint-aware optimization methods. For interpretability, Minerva also integrates feature attribution methods (such as SHAP values) to reveal which experimental parameters and chemical features most strongly influence reaction outcomes, providing chemical insight beyond predictive capability alone.
Validation and experimental applications
The authors validated the performance of Minerva extensively through simulation-based benchmark datasets and experimental applications. Benchmark datasets included virtual representations of reaction landscapes derived from high-quality experimental data, allowing comprehensive evaluation of Minerva's robustness and scalability. Through these benchmarks, Minerva consistently demonstrated superior performance in identifying optimal reaction conditions efficiently and robustly, even in scenarios with significant chemical noise and large batch sizes.
Following successful benchmarks, Minerva was applied experimentally to challenging reactions with direct pharmaceutical relevance. A notable example is the nickel-catalysed Suzuki-Miyaura coupling reaction—a promising but challenging transformation requiring non-precious metal catalysts. Using Minerva with a 96-well automated HTE platform, the authors navigated an extensive search space comprising approximately 88,000 reaction configurations. The ML-driven optimisation identified optimal reaction conditions achieving up to 76% yield and 92% selectivity, significantly outperforming traditional expert-driven approaches, which had previously struggled to identify productive conditions.
Further highlighting Minerva’s practical applicability, the authors employed their framework in pharmaceutical process chemistry for two active pharmaceutical ingredient (API) syntheses. In the first case, a nickel-catalysed Suzuki reaction was optimised, rapidly yielding multiple optimal conditions achieving greater than 95% yield and selectivity. In the second application, Minerva was employed to optimise a palladium-catalysed Buchwald–Hartwig reaction. Here, Minerva not only identified optimal conditions with high selectivity and yield (>95%) but significantly improved purity by reducing a problematic impurity previously encountered. Impressively, the ML-driven optimisation approach drastically reduced development timelines, identifying optimal process conditions in just four weeks—a substantial improvement over the six-month timeframe previously required by traditional methods.
Insights and interpretability
An important aspect of Minerva is the interpretability of its optimisation outcomes. By leveraging feature attribution methods such as SHAP (Shapley additive explanations), Minerva provides insights into the chemical parameters that drive optimisation outcomes. The authors showed how Minerva could effectively reveal unexpected yet critical chemical factors, such as particular precatalyst and ligand combinations, significantly influencing reaction performance. This interpretability feature provides chemists with a deeper understanding of reaction mechanisms and facilitates more informed decision-making during the optimisation process.
Implications and future perspectives
This study showcases a compelling advancement for chemical reaction optimisation, demonstrating the practical effectiveness of combining automation, machine learning, and chemical intuition. Minerva stands out as a robust, scalable, and highly interpretable method, effectively addressing existing challenges in optimising high-dimensional reaction spaces in industrial and pharmaceutical chemistry contexts.
The use of Minerva promises not only faster development of chemical reactions and pharmaceutical processes but also the potential to uncover novel chemical reactivity patterns that traditional approaches may overlook. While the current study demonstrates its efficacy across diverse reaction conditions and constraints, future developments could further refine Minerva’s capabilities. Incorporating dynamic, expert-guided updates to reaction search spaces during optimization could further enhance exploration and exploitation capabilities. Moreover, expanding the range and diversity of reaction classes and conditions tested could validate and extend Minerva’s applicability across even broader chemical transformations.
In conclusion, this study by Sin et al. represents a significant advancement in automated reaction optimisation, combining the powerful synergy of artificial intelligence and chemical synthesis. Minerva’s successful application across diverse chemical transformations underscores its broad applicability and potential to significantly accelerate process development in pharmaceuticals and chemical manufacturing.